Использование открытых последовательностей праймеров для разработки собственных тест-систем довольно привлекательно, так как экономит время на разработку и гарантирует какой-то результат. Однако, актуальность и применимость этой информации для решения конкретных задач необходимо проверить. В данной статье рассмотрен пример оценки специфичности литературных тест-систем для детектирования C. trachomatis по гену 16S ДНК. Этот ген довольно консервативен и может быть использован для для выявления всех представителей Chlamydiaceae.
Создание пула мишеней
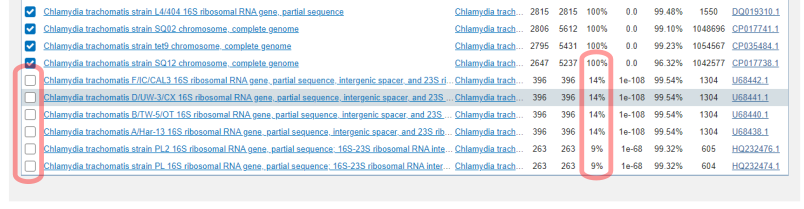
Формирование пула нуклеотидных последовательностей, которые исследователь привязал к выбранному организму или группе организмов, будет определять диагностическую специфичность будущей тест-системы. В нашем примере мы воспользуемся базой данных NCBI и сформируем запрос на нуклеотидных последовательности, которые относятся к C.trachomatis и содержат "16S" в названии. Запрос будет выглядеть вот так: "16S"[Title] AND "Chlamydia trachomatis"[porgn:__txid813]. В результате (Рси.1) мы получим 37 записей, одна из которых является проверенной ("refseq").
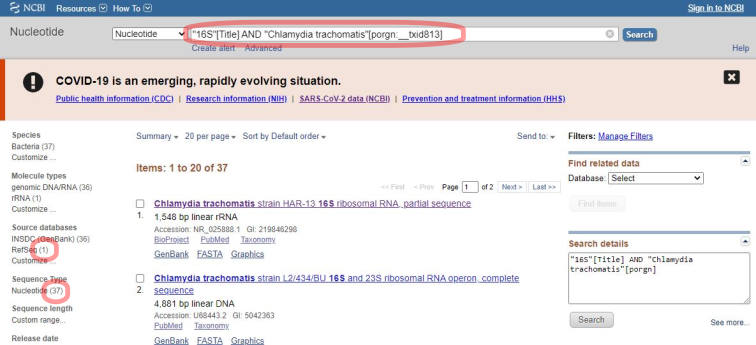
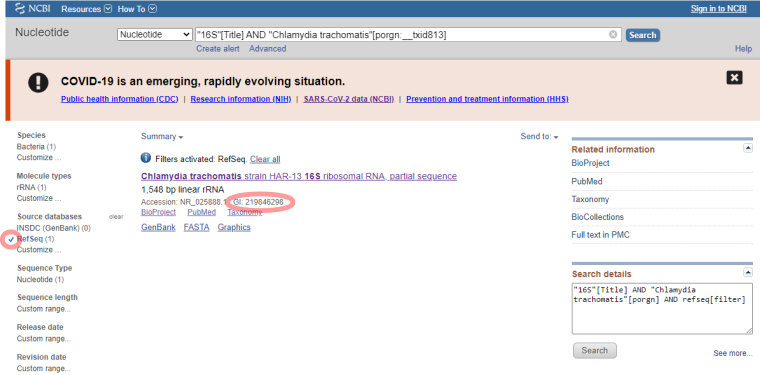
В данном запросе мы заведомо исключили записи, которые относятся к полному геному и, соответственно, не содержат "16S" в названии. Для формирования пула последовательностей мы возьмем проверенную нуклеотидную последовательность (Рис.2 Отметим "RefSeq" и скопируем номер GI) и в программе BLAST выполним поиск (Рис.3) похожих последовательностей, которые относятся к C.trachomatis.
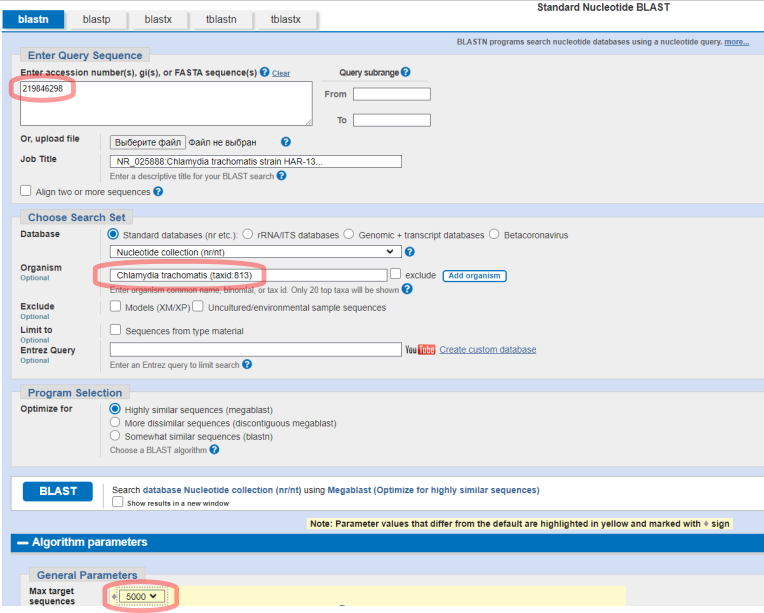
В качестве параметров мы укажем номер GI для выбранной проверенной последовательности, ограничим поиск указанием организма и увеличим предел выводимых результатов (Рис.3).
В результате мы получим ряд нуклеотидных последовательностей, которые сходны с исходной проверенной последовательностью (Рис.4). Длина совпадающего участка указана в столбце "Query Cover" и выражена в процентах относительно длины последовательности в запросе
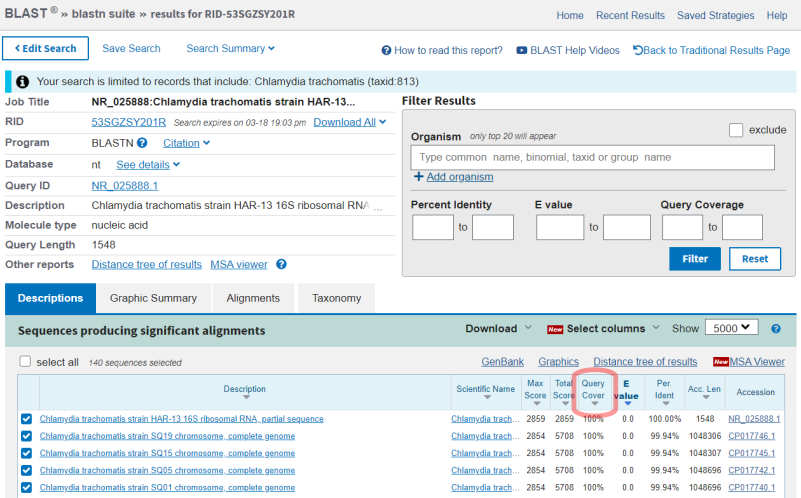
Для дальнейшей работы мы исключим записи, которые отражают совпадение по небольшому фрагменту основной последовательности (Рис.5).
Сохранить пул целевых нуклеотидных последовательностей в формате FASTA мы можем, выполнив экспорт результатов поиска (Рис.6).
Оценка специфичности праймеров
В качестве примера открытых тест-систем мы возмем праймеры из двух статей.
| № | Статья | Праймеры |
| 1 | An Q., Radcliffe G., Vassallo R., Buxton D., O'brien W., Pelletier D., Weisburg W., Klinger J., Olive M. Infection with a plasmid-free variant chlamydia related to chlamydia trachomatis identified by using multiple assays for nucleic acid detection // Journal Of Clinical Microbiology, 1992, 30(11), 2814-2821 | 5`-GAAGGCGGTTAATACCCGCTG-3` |
| 5`-GATGGGGTTGAGACCATCC-3` | ||
| 2 | Vitrenko Y., Deryabin O. A dual-target strategy for the detection of Chlamydia trachomatis by real-time PCR // Biopolymers and Cell, 2018, 34(2), 117–128 | 5`-AGTGGCGGAAGGGTTAGTAATG-3` |
| 5`-TCACATAGACTCTCCCTTAACCGA-3` |
В программе System Viewer, разработанной командой проекта PCR-Guide.ru, можно открыть файл c пулом последовательностей. Для этого нужно выбрать меню "File", затем "Open Sequence File...", в появившемся диалоге изменить формат отображаемых файлов с "*.gb" на "*.fasta" и выбрать файл с последовательностями. Далее в соответствующих полях ввести нуклеотидные последовательности праймеров и выполнить поиск (Рис.7).
Стабильная версия программы System Viewer находится в разработке.
Тестовую версию программы можно скачать тут.
С вопросами по поводу работы программы можно обращаться по почте Адрес электронной почты защищен от спам-ботов. Для просмотра адреса в вашем браузере должен быть включен Javascript.
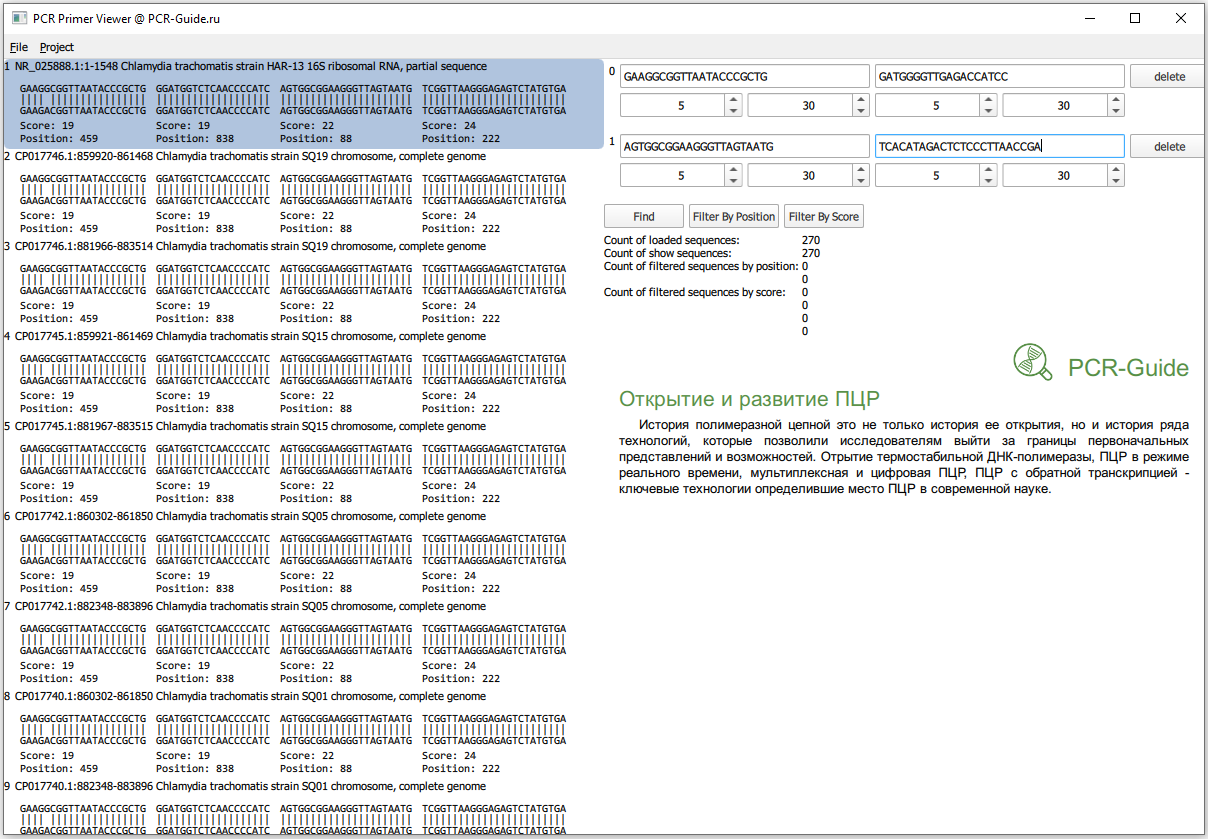
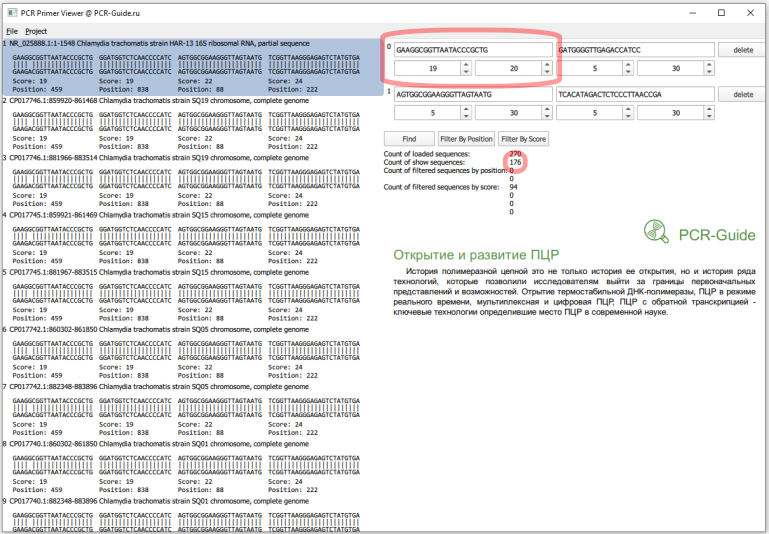
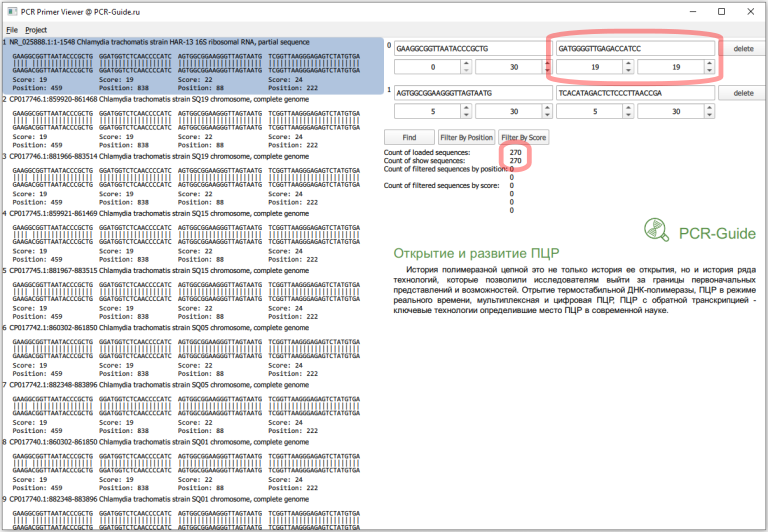
Всего получилось загружено 270 последовательностей, это больше чем посчитано в программе BLAST (140), так как часть последовательностей относятся к геному, а он содержит 2 копии гена. Сразу заметно, что прямой праймер в первом наборе не обладает 100% специфичностью и часть последовательностей содержит замену G на А в 5 нуклеотиде. Чтобы точнее оценить специфичность, необходимо изменить параметры фильтров по рейтингу. Максимальный рейтинг для праймера равен его длине, поэтому чтобы узнать количество последовательностей, которые полностью соответствуют последовательности праймеров необходимо в качестве минимальных и максимальных значений ввести длину праймеров.
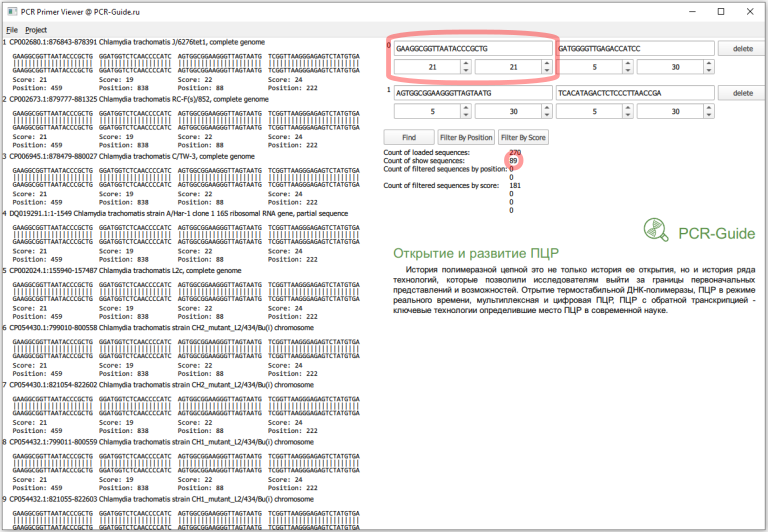
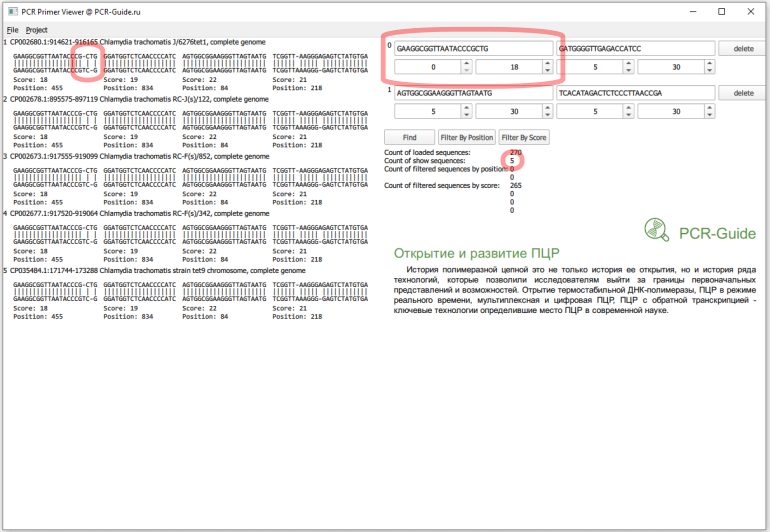
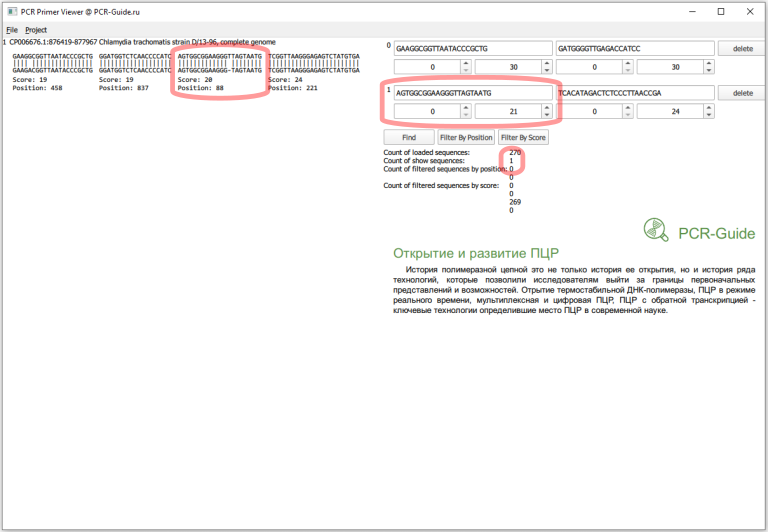
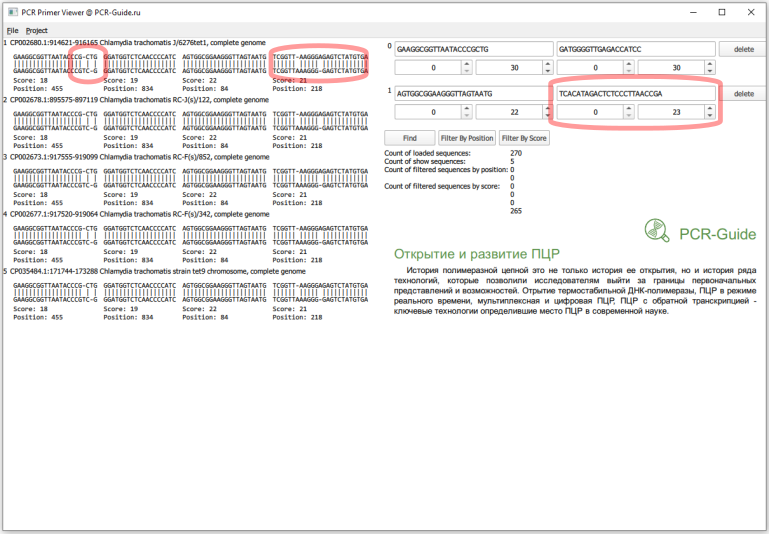
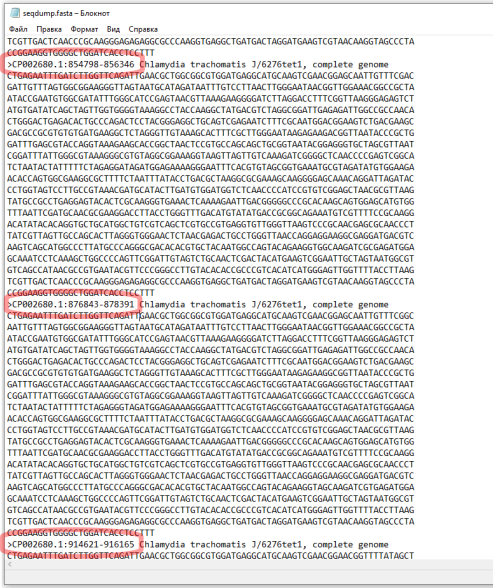
Теперь необходимо присмотреться к этим пяти последовательностям. Первая часть в названии - название локуса в базe NCBI. Если мы откроем файл *.FASTA с исходным пулом последовательностей можно найти эти последовательности и другие последовательности, которые относятся к этому локусу. Например, для локуса CP002680 существует три записи (Рис.14), которые похожи на исходную проверенную последовательности 16S. Причем, первые две хорошо соответствуют последовательностям праймеров.
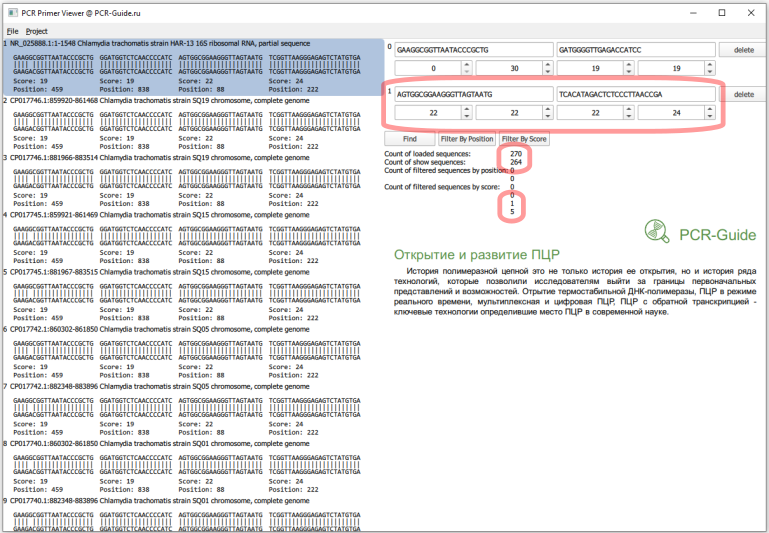
Для остальных записей ситуация аналогичная. Локусы CP002673 и CP002677 представлены в трех записях, две из которых соответствуют праймерам, CP002678 и CP035484 - в двух, одна из которых соответствует праймерам. Таким образом, эти несовпадения не повлияют на специфичности наборов праймеров, так как в геноме организма есть альтернативный участок, который будет более специфичным. То же самое с локусом CP006676, в котором есть участок, который не полностью совпадает с прямым праймером во втором наборе. Но это локус также содержит участок со 100% совпадением.
Другая ситуация с прямым праймером в первом наборе. В этом случае действительно имеет место снижение специфичности, так как геном организма не содержит альтернативного участка. Описанная замена находится на 5'-конце, поэтому не может значимо повлиять на эффективность амплификации в нормальных условиях. Но в сложных пробах, с низким содержанием ДНК и высоким содержанием мешающих компонентов эффективность может снизиться. На практике этом может выражаться в большем наличии "ненадежных", или "пограничных", результатов.